Sunday, June 29, 2008
HGP - Benefits
Benefits
The work on interpretation of genome data is still in its initial stages. It is anticipated that detailed knowledge of the human genome will provide new avenues for advances in medicine and biotechnology. Clear practical results of the project emerged even before the work was finished. For example, a number of companies, such as Myriad Genetics started offering easy ways to administer genetic tests that can show predisposition to a variety of illnesses, including breast cancer, disorders of hemostasis, cystic fibrosis, liver diseases and many others.
Also, the etiologies for cancers, Alzheimer's disease and other areas of clinical interest are considered likely to benefit from genome information and possibly may lead in the long term to significant advances in their management.
There are also many tangible benefits for biological scientists. For example, a researcher investigating a certain form of cancer may have narrowed down his/her search to a particular gene. By visiting the human genome database on the world wide web, this researcher can examine what other scientists have written about this gene, including (potentially) the three-dimensional structure of its product, its function(s), its evolutionary relationships to other human genes, or to genes in mice or yeast or fruit flies, possible detrimental mutations, interactions with other genes, body tissues in which this gene is activated, diseases associated with this gene or other datatypes.
Further, deeper understanding of the disease processes at the level of molecular biology may determine new therapeutic procedures. Given the established importance of DNA in molecular biology and its central role in determining the fundamental operation of cellular processes, it is likely that expanded knowledge in this area will facilitate medical advances in numerous areas of clinical interest that may not have been possible without them.
The analysis of similarities between DNA sequences from different organisms is also opening new avenues in the study of the theory of evolution. In many cases, evolutionary questions can now be framed in terms of molecular biology; indeed, many major evolutionary milestones (the emergence of the ribosome and organelles, the development of embryos with body plans, the vertebrate immune system) can be related to the molecular level.
Many questions about the similarities and differences between humans and our closest relatives (the primates, and indeed the other mammals) are expected to be illuminated by the data from this project.
The Human Genome Diversity Project (HGDP), spinoff research aimed at mapping the DNA that varies between human ethnic groups, which was rumored to have been halted, actually did continue and to date has yielded new conclusions.[citation needed] In the future, HGDP could possibly expose new data in disease surveillance, human development and anthropology. HGDP could unlock secrets behind and create new strategies for managing the vulnerability of ethnic groups to certain diseases (see race in biomedicine). It could also show how human populations have adapted to these vulnerabilities.
The Human Genome Project
The Human Genome Project
The Human Genome Project (HGP) was an international scientific research project with a primary goal to determine the sequence of chemical base pairs, which make up DNA and to identify the approximately 25,000 genes of the human genome from both a physical and functional standpoint.
The project began in 1990 initially headed by James D. Watson at the U.S. National Institutes of Health. A working draft of the genome was released in 2000 and a complete one in 2003, with further analysis still being published. A parallel project was conducted by the private company Celera Genomics. Most of the sequencing was performed in universities and research centers from the United States, Canada and Great Britain. The mapping of human genes is an important step in the development of medicines and other aspects of health care.
While the objective of the Human Genome Project is to understand the genetic makeup of the human species, the project also has focused on several other nonhuman organisms such as E. coli, the fruit fly, and the laboratory mouse. It remains one of the largest single investigational projects in modern science.[citation needed].
The HGP originally aimed to map the nucleotides contained in a haploid reference human genome (more than three billion). Several groups have announced efforts to extend this to diploid human genomes including the International HapMap Project, Applied Biosystems, Perlegen, Illumina, JCVI, Personal Genome Project, and Roche-454.
The "genome" of any given individual (except for identical twins and cloned animals) is unique; mapping "the human genome" involves sequencing multiple variations of each gene. The project did not study the entire DNA found in human cells; some heterochromatic areas (about 8% of the total) remain un-sequenced.
History
In 1976, the genome of the virus Bacteriophage MS2 was the first complete genome to be determined, by Walter Fiers and his team at the University of Ghent (Ghent, Belgium). The idea for the shotgun technique came from the use of an algorithm that combined sequence information from many small fragments of DNA to reconstruct a genome. This technique was pioneered by Frederick Sanger to sequence the genome of the Phage Φ-X174, a virus that primarily infects bacteria (bacteriophage) that was the first fully sequenced genome (DNA-sequence) in 1977.
The technique was called shotgun sequencing because the genome was broken into millions of pieces as if it had been blasted with a shotgun. In order to scale up the method, both the sequencing and genome assembly had to be automated, as they were in the 1980s.
Those techniques were shown applicable to sequencing of the first free-living bacterial genome (1.8 million base pairs) of Haemophilus influenzae in 1995 and the first animal genome (~100 Mbp) It involved the use of automated sequencers, longer individual sequences using approximately 500 base pairs at that time. Paired sequences separated by a fixed distance of around 2000 base pairs which were critical elements enabling the development of the first genome assembly programs for reconstruction of large regions of genomes (aka 'contigs').
Three years later, in 1998, the announcement by the newly-formed Celera Genomics that it would scale up the shotgun sequencing method to the human genome was greeted with skepticism in some circles. The shotgun technique breaks the DNA into fragments of various sizes, ranging from 2,000 to 300,000 base pairs in length, forming what is called a DNA "library". Using an automated DNA sequencer the DNA is read in 800bp lengths from both ends of each fragment. Using a complex genome assembly algorithm and a supercomputer, the pieces are combined and the genome can be reconstructed from the millions of short, 800 base pair fragments. The success of both the public and privately funded effort hinged upon a new, more highly automated capillary DNA sequencing machine, called the Applied Biosystems 3700, that ran the DNA sequences through an extremely fine capillary tube rather than a flat gel. Even more critical was the development of a new, larger-scale genome assembly program, which could handle the 30-50 million sequences that would be required to sequence the entire human genome with this method.
At the time, such a program did not exist. One of the first major projects at Celera Genomics was the development of this assembler, which was written in parallel with the construction of a large, highly automated genome sequencing factory. Development of the assembler was led by Brian Ramos. The first version of this assembler was demonstrated in 2000, when the Celera team joined forces with Professor Gerald Rubin to sequence the fruit fly Drosophila melanogaster using the whole-genome shotgun method. At 130 million base pairs, it was at least 10 times larger than any genome previously shotgun assembled. One year later, the Celera team published their assembly of the three billion base pair human genome.
ELISA - Applications
Applications
Because the ELISA can be performed to evaluate either the presence of antigen or the presence of antibody in a sample, it is a useful tool both for determining serum antibody concentrations (such as with the HIV test or West Nile Virus) and also for detecting the presence of antigen.
It has also found applications in the food industry in detecting potential food allergens such as milk, peanuts, walnuts, almonds, and eggs. ELISA can also be used in toxicology as a rapid presumptive screen for certain classes of drugs.
The ELISA test, or the enzyme immunoassay (EIA), was the first screening test commonly employed for HIV. It has a high sensitivity. In an ELISA test, a person's serum is diluted 400-fold and applied to a plate to which HIV antigens have been attached. If antibodies to HIV are present in the serum, they may bind to these HIV antigens. The plate is then washed to remove all other components of the serum. A specially prepared "secondary antibody" — an antibody that binds to other antibodies — is then applied to the plate, followed by another wash.
This secondary antibody is chemically linked in advance to an enzyme. Thus the plate will contain enzyme in proportion to the amount of secondary antibody bound to the plate. A substrate for the enzyme is applied, and catalysis by the enzyme leads to a change in color or fluorescence. ELISA results are reported as a number; the most controversial aspect of this test is determining the "cut-off" point between a positive and negative result.
One method of determining a cut-off point is by comparison with a known standard. For example, if an ELISA test will be used in workplace drug screening, a cut-off concentration (e.g., 50 ng/mL of drug) will be established and a sample will be prepared that contains that concentration of analyte. Unknowns that generate a signal that is stronger than the known sample are called "positive"; those that generate weaker signal are called "negative."
ELISA
ELISA (Enzyme-Linked ImmunoSorbant Assay)
The purpose of an ELISA is to determine if a particular protein is present in a sample and if so, how much. There are two main variations on this method: you can determine how much antibody is in a sample, or you can determine how much protein is bound by an antibody.
The distinction is whether you are trying to quantify an antibody or some other protein. In this example, we will use an ELISA to determine how much of a particular antibody is present in an individuals blood.

ELISAs are performed in 96-well plates which permits high throughput results. The bottom of each well is coated with a protein to which will bind the antibody you want to measure. Whole blood is allowed to clot and the cells are centrifuged out to obtain the clear serum with antibodies (called primary antibodies). The serum is incubated in a well, and each well contains a different serum (see figure below). A positive control serum and a negative control serum would be included among the 96 samples being tested.
After some time, the serum is removed and weakly adherent antibodies are washed off with a series of buffer rinses. To detect the bound antibodies, a secondary antibody is added to each well. The secondary antibody would bind to all human antibodies and is typically produced in a rodent. Attached to the secondary antibody is an enzyme such as peroxidase or alkaline phosphatase. These enzymes can metabolize colorless substrates (sometimes called chromagens) into colored products. After an incubation period, the secondary antibody solution is removed and loosely adherent ones are washed off as before. The final step is the addition the enzyme substrate and the production of colored product in wells with secondary antibodies bound.
When the enzyme reaction is complete, the entire plate is placed into a plate reader and the optical density (i.e. the amount of colored product) is determined for each well. The amount of color produced is proportional to the amount of primary antibody bound to the proteins on the bottom of the wells.
Agarose Gel Electrophoresis
Agarose Gel Electrophoresis
Introduction:
Agarose gel electrophoresis is an easy way to separate DNA fragments by their sizes and visualize them. It is a common diagnostic procedure used in molecular biological labs.
Electrophoresis:
The technique of electrophoresis is based on the fact that DNA is negatively charged at neutral pH due to its phosphate backbone. For this reason, when an electrical potential is placed on the DNA it will move toward the positive pole:

Dig. 1
The rate at which the DNA will move toward the positive pole is slowed by making the DNA move through an agarose gel. This is a buffer solution (which maintains the proper pH and salt concentration) with 0.75% to 2.0% agarose added. The agarose forms a porous lattice in the buffer solution and the DNA must slip through the holes in the lattice in order to move toward the positive pole. This slows the molecule down. Larger molecules will be slowed down more than smaller molecules, since the smaller molecules can fit through the holes easier. As a result, a mixture of large and small fragments of DNA that has been run through an agarose gel will be separated by size. This is a graphic representation of an agarose gel made by "running" DNA molecular weight markers, an isolated plasmid, and the same plasmid after linearization with a restriction enzyme:
These gels are visualized on a U.V. trans-illuminator by staining the DNA with a fluorescent dye (ethidium bromide). The DNA molecular weight marker is a set of DNA fragments of known molecular sizes that are used as a standard to determine the sizes of your unknown fragments.
If you click on the figure you will see a short movie that simulates the movement of the DNA bands through the gel. When looking at the video, note that bands of a low molecular weight move very quickly through the gel while high molecular weight bands move very slowly.
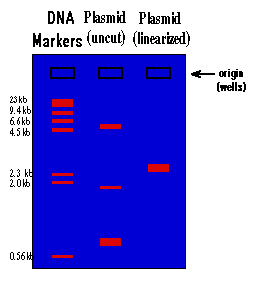
Dig. 2
Interpretation:
Much information can be derived from this gel. As you read the text below,
1.) By looking at the migration of the DNA molecular weight standards, you can tell that the migration of DNA through an agarose gel is not linear with respect to size. If you graphed the distance traveled vs. the molecular weight of the fragment, you would see that there is a logarithmic relationship (i.e. small fragments travel much faster than large fragments).
2.) You can see that there is a big difference between the way a plasmid as isolated from the alkaline lysis prep will run vs. this same plasmid after it is cut with a restriction enzyme and linearized. This is because the plasmid will be found in many different supercoiled forms in the bacteria. When you isolate plasmid from a bacterial culture, you isolate all the different supercoiled forms of the plasmid, and each will migrate differently on the gel, giving you three major bands and many minor bands. When this mixture of supercoiled plasmids is cut with a restriction enzyme, the different forms linearize and unwind. As a result they all become identical and run at the same rate, and you see only one band on the gel.
3.) The molecular size of an unknown piece of DNA can be estimated by comparison of the distance that it travels with that of the molecular weight standards. This is only true for linear DNA. None of the supercoiled forms will migrate at a rate relative to linear DNA, which means that you can't use the DNA markers to estimate the molecular weight of a circular DNA molecule. To estimate the molecular weight of a plasmid, you must first linearize it. By looking at the gel above, the molecular size of the plasmid can be estimated at approximately 3.0 kilobases (kb). A more accurate estimate can be found by graphing the molecular weight of the standards (in base pairs) vs. the distance traveled on semi-log paper and using this graph to determine the molecular weight of the unknown. You will do this at the end of this experiment. Molecular size is the most important information derived from the agarose gel and the usual reason for running a gel.
In this experiment, you will linearize the plasmid that you isolated last week with a restriction enzyme. Then you will run this linearized plasmid on an agarose gel with the uncut version and a DNA marker to determine the size of your plasmid + insert, which will give you an estimate of the size of your insert.
Procedure:
1.) Put together the following reaction mixture for the restriction digestion:
14.5 ul water
2.0 ul 10X Rest. Enzyme buffer
3.0 ul plasmid DNA solution (from last week)
0.5 ul Restriction Enzyme (eg., HindIII)
20.0 ul Total
Add the enzyme last, and always keep it on ice. The enzyme you will use will depend on the plasmid that you have, and will be told to you during class. 0.5 ul can't be measured with your pipetman. You must estimate it by the way it will look in the pipet tip (instruction will be given in class). Be sure to use a clean tip when taking the enzyme out of the tube. Put this reaction at 37oC for 45 minutes.
2.) When the digestion is complete, prepare to load the gel. In a new tube, place 17.0 ul of H2O and 3.0 ul of uncut plasmid DNA. Add 2.0 ul dye to each of the three sample tubes (DNA markers, uncut plasmid, and digested plasmid). Load 20.0 ul of DNA marker in to one well of the gel. Do this by sucking the solution into the pipet tip, placing the tip in the top of the well, and gently expelling the liquid into the well. The dye buffer in the DNA marker and samples contains glycerol which makes it more dense than H2O. This will cause the liquid to sink to the bottom of the well. Load 20.0 ul of the uncut plasmid and the restriction digestion.
3.) Turn on the power supply and electrophorese the samples at 110 V (Warning- be careful of the high voltage or you will be set down on your butt dramatically.) Electrophorese the samples until the dark blue dye is about 2 cm from the bottom of the gel
4.) Stain the gel by incubating it for 8 min in an ethidium bromide solution.
WARNING!
Ethidium bromide is very carcinogenic. Handle this gel only while wearing gloves. Never put unprotected fingers in the gel buffer solution.
5.) I will move the gel onto the U.V. trans illuminator and take a picture of it.
6.) I will denature the ethidium bromide by placing the gel in potassium permanganate solution for 5 minutes, then discard it.
Calculations:
On a piece of semi-log graph paper, plot the log of the molecular weight each standard vs. distance traveled from the well (based on measurements made from the picture of the gel). The sizes of the molecular weight standards may be different than in figure 2 and will be given to you during lab. Draw a line connecting the points (although the line won't be linear). From this you should be able to determine the molecular size of the linearized plasmid. Note the difference between the way the linearized DNA ran and the way the uncut plasmid appears. If you have multiple bands in the digested plasmid lane, see if some of them are of the same size as bands in the uncut lane; you may have incomplete digestion of your plasmid due to a sub-optimal purification (i.e., a dirty plasmid prep). Since each of the vector plasmids (without the insert) is 3.0 kb, you can determine the size of the insert in your plasmid by subtracting 3.0 from the size estimated from the gel.
Animal Biotechnology
Animal Biotechnology
Animal biotechnology is the application of scientific and engineering principles to the processing or production of materials by animals or aquatic species to provide goods and services (NRC 2003). Examples of animal biotechnology include generation of transgenic animals or transgenic fish (animals or fish with one or more genes introduced by human intervention), using gene knockout technology to generate animals in which a specific gene has been inactivated, production of nearly identical animals by somatic cell nuclear transfer (also referred to as clones), or production of infertile aquatic species.
Transgenics
Since the early 1980s, methods have been developed and refined to generate transgenic animals or transgenic aquatic species. For example, transgenic livestock and transgenic aquatic species have been generated with increased growth rates, enhanced lean muscle mass, enhanced resistance to disease or improved use of dietary phosphorous to lessen the environmental impacts of animal manure.

Transgenic poultry, swine, goats, and cattle also have been produced that generate large quantities of human proteins in eggs, milk, blood, or urine, with the goal of using these products as human pharmaceuticals. Examples of human pharmaceutical proteins include enzymes, clotting factors, albumin, and antibodies. The major factor limiting widespread use of transgenic animals in agricultural production systems is the relatively inefficient rate (success rate less than 10 percent) of production of transgenic animals. CSREES has supported research projects to generate transgenic animals or transgenic aquatic species with enhanced production or health traits.
Gene Knockout Technology
Animal biotechnology also can knock out or inactivate a specific gene. Knockout technology creates a possible source of replacement organs for humans. The process of transplanting cells, tissues, or organs from one species to another is referred to as “xenotransplantation.” Currently, the pig is the major animal being considered as a xenotransplant donor to humans. Unfortunately, pig cells and human cells are not immunologically compatible. Pig cells express a carbohydrate epitope (alpha1, 3 galactose) on their surface that is not normally found on human cells.
Humans will generate antibodies to this epitope, which will result in acute rejection of the xenograft. Genetic engineering is used to knock out or inactivate the pig gene (alpha1, 3 galactosyl transferase) that attaches this carbohydrate epitope on pig cells. Other examples of knockout technology in animals include inactivation of the prion-related peptide (PRP) gene that may generate animals resistant to diseases associated with prions (bovine spongiform encephalopathy [BSE], Creutzfeldt-Jakob Disease [CJD], scrapie, etc.). Most of the funding for these types of projects is conducted by private companies or in academic laboratories supported by the National Institutes of Health. Research projects designed to provide basic information regarding mechanisms associated with gene knockout technology are supported by CSREES.
Somatic Cell Nuclear Transfer
Another application of animal biotechnology is the use of somatic cell nuclear transfer to produce multiple copies of animals that are nearly identical copies of other animals (transgenic animals, genetically superior animals, or animals that produce high quantities of milk or have some other desirable trait, etc.).

This process has been referred to as cloning. To date, somatic cell nuclear transfer has been used to clone cattle, sheep, pigs, goats, horses, mules, cats, rats, and mice. The technique involves culturing somatic cells from an appropriate tissue (fibroblasts) from the animal to be cloned. Nuclei from the cultured somatic cells are then microinjected into an enucleated oocyte obtained from another individual of the same or a closely related species.
Through a process that is not yet understood, the nucleus from the somatic cell is reprogrammed to a pattern of gene expression suitable for directing normal development of the embryo. After further culture and development in vitro, the embryos are transferred to a recipient female and ultimately will result in the birth of live offspring. The success rate for propagating animals by nuclear transfer is often less than 10 percent and depends on many factors, including the species, source of the recipient ova, cell type of the donor nuclei, treatment of donor cells prior to nuclear transfer, the techniques employed for nuclear transfer, etc. CSREES has supported research projects to obtain a better understanding of the basic cellular mechanisms associated with nuclear reprogramming.
Production of Infertile Aquatic Species. In aquaculture production systems, some species are not indigenous to a given area and can pose an ecological risk to native species should the foreign species escape confinement and enter the natural ecosystem. Generation of large populations of sterile fish or mollusks is one potential solution to this problem. Techniques have been developed to alter the chromosome complement to render individual fish and mollusks infertile. For example, triploid individuals (with three, instead of two, sets of chromosomes) have been generated by using various procedures to interfere with the final step in meiosis (extrusion of the second polar body). Timed application of high or low temperatures, various chemicals, or high hydrostatic pressure to newly fertilized eggs has been effective in producing triploid individuals. At a later time, the first cell division of the zygote can be suppressed to produce a fertile tetraploid individual (four sets of chromosomes). Tetraploids can then be mated with normal diploids to produce large numbers of infertile triploids. Unfortunately, in a commercial production system, it is often difficult to obtain sterilization of 100 percent of the individuals; thus, alternative methods are needed to ensure reproductive confinement of transgenic fish. Another technique that is being developed for finfish is to farm monosex fish stocks. Monosex populations can be produced by gender reversal and progeny testing to identify XX males for producing all female stocks or YY males for producing all male stocks. CSREES has supported research projects to alter the chromosome content or produce monosex populations of genetically engineered fish or mollusks.
As with any new technology, animal biotechnology faces a variety of uncertainties, safety issues and potential risks. For example, concerns have been raised regarding: the use of unnecessary genes in constructs used to generate transgenic animals, the use of vectors with the potential to be transferred or to otherwise contribute sequences to other organisms, the potential effects of genetically modified animals on the environment, the effects of the biotechnology on the welfare of the animal, and potential human health and food safety concerns for meat or animal products derived from animal biotechnology. Before animal biotechnology will be used widely by animal agriculture production systems, additional research will be needed to determine if the benefits of animal biotechnology outweigh these potential risks.
The USDA Biotechnology Risk Assessment Grants program supports environmental risk assessment research projects on genetically engineered animals. In addition, the NRI Animal Protection program supports research projects to determine the effects of genetic modification on the health and well-being of the animal.
Advances in animal biotechnology have been facilitated by recent progress in sequencing and analyzing animal genomes, identification of molecular markers (microsatellites, expressed sequence tags [ESTs], quantitative trait loci [QTLs], etc.) and a better understanding of the mechanisms that regulate gene expression.
Genetic Engineering Advantages & Disadvantages
Genetic Engineering Advantages & Disadvantages
During the latter stage stages of the 20th century, man harnessed the power of the atom, and not long after, soon realised the power of genes. Genetic engineering is going to become a very mainstream part of our lives sooner or later, because there are so many possibilities advantages (and disadvantages) involved.
Here are just some of the advantages :
1) Disease could be prevented by detecting people/plants/animals that are genetically prone to certain hereditary diseases, and preparing for the inevitable. Also, infectious diseases can be treated by implanting genes that code for antiviral proteins specific to each antigen.
2) Animals and plants can be 'tailor made' to show desirable characteristics. Genes could also be manipulated in trees for example, to absorb more CO2 and reduce the threat of global warming.
3) Genetic Engineering could increase genetic diversity, and produce more variant alleles which could also be crossed over and implanted into other species. It is possible to alter the genetics of wheat plants to grow insulin for example.
Of course there are two sides to the coin, here are some possible eventualities and disadvantages.
1) Nature is an extremely complex inter-related chain consisting of many species linked in the food chain. Some scientists believe that introducing genetically modified genes may have an irreversible effect with consequences yet unknown.
2) Genetic engineering borderlines on many moral issues, particularly involving religion, which questions whether man has the right to manipulate the laws and course of nature.
Genetic engineering may be one of the greatest breakthroughs in recent history alongside the discovery of the atom and space flight, however, with the above eventualities and facts above in hand, governments have produced legislation to control what sort of experiments are done involving genetic engineering. In the UK there are strict laws prohibiting any experiments involving the cloning of humans. However, over the years here are some of the experimental 'breakthroughs' made possible by genetic engineering.
1) At the Roslin Institute in Scotland, scientists successfully cloned an exact copy of a sheep, named 'Dolly'. This was the first successful cloning of an animal, and most likely the first occurrence of two organisms being genetically identical. Note : Recently the sheep's health has deteriorated detrimentally
2) Scientists successfully manipulated the genetic sequence of a rat to grow a human ear on its back. (Unusual, but for the purpose of reproducing human organs for medical purposes).
3) Most controversially, and maybe due to more liberal laws, an American scientist is currently conducting tests to clone himself.
Genetic engineering has been impossible until recent times due to the complex and microscopic nature of DNA and its component nucleotides. Through progressive studies, more and more in this area is being made possible, with the above examples only showing some of the potential that genetic engineering shows.
For us to understand chromosomes and DNA more clearly, they can be mapped for future reference. More simplistic organisms such as fruit fly (Drosophila) have been chromosome mapped due to their simplistic nature meaning they will require less genes to operate. At present, a task named the Human Genome Project is mapping the human genome, and should be completed in the next ten years.
The process of genetic engineering involves splicing an area of a chromosome, a gene, that controls a certain characteristic of the body. The enzyme endonuclease is used to split a DNA sequence and split the gene from the rest of the chromosome. For example, this gene may be programmed to produce an antiviral protein.
This gene is removed and can be placed into another organism. For example, it can be placed into a bacteria, where it is sealed into the DNA chain using ligase. When the chromosome is once again sealed, the bacteria is now effectively re-programmed to replicate this new antiviral protein. The bacteria can continue to live a healthy life, though genetic engineering and human intervention has actively manipulated what the bacteria actually is. No doubt there are advantages and disadvantages, and this whole subject area will become more prominent over time.
Genetic Engineering Techniques
Genetic Engineering Techniques
There are a number of techniques for moving genes artificially into recipient organisms. The oldest of these is called recombinant DNA, a technique that relies on biological vectors like plasmids or viruses. Other newer gene transfer techniques are electro- and chemical poration, microinjection, and bioballistics.
Recombinant DNA
Recombinant DNA techniques use biological vectors like plasmids and viruses to carry foreign genes into cells. Plasmids are small circular pieces of genetic material found in bacteria that have the ability to cross species boundaries. The circles can be broken and new genetic material added to them. Plasmids augmented with new genetic material can move across microbial cell boundaries and place the new genetic material next to the bacterium's own genes. Often the bacteria will take up the gene and begin to produce the protein for which the gene codes. Where the new gene codes for insulin, for example, the bacterium will begin to produce insulin along with its other gene products. A large vat of bacteria engineered to produce insulin can then become a sort of pharmaceutical factory.
Viruses can also act as vectors in genetic engineering. Viruses are infectious particles that contain genetic material to which a new gene can be added. The virus can carry the new gene into a recipient cell in the process of infecting that cell. The virus can also be disabled so that while it can carry a new gene into a cell, it cannot redirect the cell's genetic machines to make thousands of copies of itself.
Microinjection
Other methods do not rely on biological vectors like plasmids and viruses. One of these is called microinjection and involves simply injecting genetic material containing the new gene into the recipient cell. Where the cell is large enough, as many plant and animal cells are, the injection can be done with a fine-tipped glass needle. Somehow the injected genes find the host cell genes and incorporate themselves among them.

Electro- and Chemical Poration
Other methods for direct gene transfer involve creating pores or holes in the cell membrane to allow entry of the new genes. This can be done by bathing cells in solutions of special chemicals--so-called chemical poration--or subjecting cells to a weak electric current--so-called electroporation.
Bioballistics
Finally, there are so-called projectile methods that use metal slivers to deliver the genetic material to the interior of the cell. The small slivers (much smaller than the diameter of the target cell) are coated with genetic material. One projectile method, called bioballistics, propels the coated slivers into the cell using a shot gun. A perforated metal plate stops the shell cartridge, but allows the slivers to pass through and into the living cells on the other side. Once in the cell, the genetic material is transported to the nucleus where it is incorporated among the host genes.
There are a number of techniques for moving genes artificially into recipient organisms. The oldest of these is called recombinant DNA, a technique that relies on biological vectors like plasmids or viruses. Other newer gene transfer techniques are electro- and chemical poration, microinjection, and bioballistics.
Recombinant DNA
Recombinant DNA techniques use biological vectors like plasmids and viruses to carry foreign genes into cells. Plasmids are small circular pieces of genetic material found in bacteria that have the ability to cross species boundaries. The circles can be broken and new genetic material added to them. Plasmids augmented with new genetic material can move across microbial cell boundaries and place the new genetic material next to the bacterium's own genes. Often the bacteria will take up the gene and begin to produce the protein for which the gene codes. Where the new gene codes for insulin, for example, the bacterium will begin to produce insulin along with its other gene products. A large vat of bacteria engineered to produce insulin can then become a sort of pharmaceutical factory.
Viruses can also act as vectors in genetic engineering. Viruses are infectious particles that contain genetic material to which a new gene can be added. The virus can carry the new gene into a recipient cell in the process of infecting that cell. The virus can also be disabled so that while it can carry a new gene into a cell, it cannot redirect the cell's genetic machines to make thousands of copies of itself.
Microinjection
Other methods do not rely on biological vectors like plasmids and viruses. One of these is called microinjection and involves simply injecting genetic material containing the new gene into the recipient cell. Where the cell is large enough, as many plant and animal cells are, the injection can be done with a fine-tipped glass needle. Somehow the injected genes find the host cell genes and incorporate themselves among them.
Electro- and Chemical Poration
Other methods for direct gene transfer involve creating pores or holes in the cell membrane to allow entry of the new genes. This can be done by bathing cells in solutions of special chemicals--so-called chemical poration--or subjecting cells to a weak electric current--so-called electroporation.
Bioballistics
Finally, there are so-called projectile methods that use metal slivers to deliver the genetic material to the interior of the cell. The small slivers (much smaller than the diameter of the target cell) are coated with genetic material. One projectile method, called bioballistics, propels the coated slivers into the cell using a shot gun. A perforated metal plate stops the shell cartridge, but allows the slivers to pass through and into the living cells on the other side. Once in the cell, the genetic material is transported to the nucleus where it is incorporated among the host genes.
What Is Genetic Engineering?
What Is Genetic Engineering?
Genetic engineering refers to a set of technologies that are being used to change the genetic makeup of cells and move genes across species boundaries to produce novel organisms. The techniques involve highly sophisticated manipulations of genetic material and other biologically important chemicals.
Genes are the chemical blueprints that determine an organism's traits. Moving genes from one organism to another transfers those traits. Through genetic engineering, organisms are given new combinations of genes—and therefore new combinations of traits—that do not occur in nature and, indeed, cannot be developed by natural means. Such an artificial technology is radically different from traditional plant and animal breeding.
Novel organisms
Nature can produce organisms with new gene combinations through sexual reproduction. A brown cow bred to a yellow cow may produce a calf of a completely new color. But reproductive mechanisms limit the number of new combinations. Cows must breed with other cows (or very near relatives). A breeder who wants a purple cow would be able to breed toward one only if the necessary purple genes were available somewhere in a cow or a near relative to cows. A genetic engineer has no such restriction. If purple genes are available anywhere in nature—in a sea urchin or an iris—those genes could be used in attempts to produce purple cows. This unprecedented ability to shuffle genes means that genetic engineers can concoct gene combinations that would never be found in nature.
New risks
Contrary to the arguments made by some proponents, genetic engineering is far from being a minor extension of existing breeding technologies. It is a radically new technology for altering the traits of living organisms by inserting genetic material that has been manipulated by artificial means. Because of this, genetic engineering may one day encompass the routine addition of novel genes that have been wholly synthesized in the laboratory.
Novel organisms bring novel risks, however, as well as the desired benefits. These risks must be carefully assessed to make sure that all effects—both desired and unintended—are benign. UCS advocates caution, examination of alternatives, and careful case-by-case evaluation of genetic enginering applications within an overall framework that seeks to move agricultural systems of food production toward sustainability.
Traditional biotechnology Vs New Biotechnology.
Traditional Biotechnologies
A prime example of traditional genetic biotechnologies is selective breeding of plants and animals. The rudiments of selecting plants and animals with desirable traits and breeding them under controlled conditions probably go back to the dawn of civilization, but the expansion of knowledge about genetics and biology in this century has developed selective breeding into a powerful and sophisticated technology. New molecular approaches like marker-assisted breeding (which enhances traditional breeding through knowledge of which cultivars or breeds carry which trait) promise to enhance these approaches even further.
Traditional breeding technologies have been immensely successful, and indeed are largely responsible for the high yields associated with contemporary agriculture. These technologies should not be considered passé or out of date. For multigene traits like intrinsic yield and drought resistance, they surpass genetic engineering. This is because selective breeding operates on whole organisms—complete sets of coordinated genes—while genetic engineering is restricted to three or four gene transfers with little control over where the new genes are inserted. For the most important agronomic traits, traditional breeding remains the technology of choice.
Other traditional nongenetic biotechnologies include the fermentation of microorganisms to produce wine, beer, and cheese. Industry also uses microorganisms to produce various products such as enzymes for use in laundry detergents. In an effort to find microorganisms that produce large amounts of enzymes, scientists sometimes treat a batch of organisms with radiation or chemicals to randomly produce genetic alternations. The process, called mutagenesis, produces numerous genetic changes in the bacteria, among which might be a few that produce more of the desired product.
New Biotechnologies
Many new biotechnologies do not involve modifications of traits passed on to the next generation. A good example is monoclonal antibodies (highly specific preparations of antibodies that bind to a single site on a protein), which have many diagnostic applications, including home pregnancy testing kits. Many biotechnology companies are engaged in these sophisticated, but noncontroversial, technologies.
By contrast, mammalian cloning is a new biotechnology that does not involve gene modification, but is nevertheless highly controversial. Cloning reproduces adult mammals by transplanting a nucleus from adult cells into an egg from which the nucleus has been removed and allowing the egg to develop in a surrogate manner. The resulting individuals are as similar to the adults from which the nuclei were taken as identical twins are to one another. Although this procedure has profound implications for human reproduction, it does not modify specific traits of an individual, but rather transfers a whole nucleus containing a complete set of genetic information.
The new technology that can affect future generations is genetic engineering, a technology based on the artificial manipulation and transfer of genetic material. This technology can move genes and the traits they dictate across natural boundaries—from one type of plant to another, from one type of animal to another, and even from a plant to an animal or an animal to a plant. Cells modified by these techniques pass the new genes and traits on to their offspring. Genetic engineering can apply to any kind of living organism from microorganisms to humans.
Genetic engineering can be applied to humans to replace or supplement defective genes. Where engineering is intended to cure disease, it is called gene therapy. Potential applications that are not related to disease, such as the modification of traits like height, are sometimes called genetic enhancement. Currently, most genetic engineering of humans is done on nonreproductive or somatic cells, like those from bone marrow. The effects of this somatic cell gene therapy are confined to the treated individual. By contrast, germ line gene therapy would modify reproductive cells, so that the modification could be passed on to future generations.
Historical Events in Biotechnology
Historical Events in Biotechnology
BC
1750 The Sumerians brew beer.
500 The Chinese use moldy soybean curds as an antibiotic to treat boils.
250 The Greeks practice crop rotation to maximize soil fertility.
100 Powdered chrysanthemum is used in China as an insecticide.
AD: Before the 20th Century
1590 The microscope is invented by Janssen.
1663 Cells are first described by Hooke.
1675 Leeuwenhoek discovers protozoa and bacteria.
1797 Jenner inoculates a child with a viral vaccine to protect him from smallpox.
1802 The word "biology" first appears.
1824 Dutrochet discovers that tissue is composed of living cells.
1830 Proteins are discovered.
1833 The cell nucleus is discovered.
The first enzymes are isolated.
1855 The Escherichia coli bacterium is discovered. It later becomes a major research, development, and production tool for biotechnology.
Pasteur begins working with yeast, eventually proving they are living organisms.
1863 Mendel, in his study of peas, discovers that traits were transmitted from parents to progeny by discrete, independent units, later called genes. His observations lay the groundwork for the field of genetics.
1869 Miescher discovers DNA in the sperm of trout.
1877 A technique for staining and identifying bacteria is developed by Koch.
1878 The first centrifuge is developed by Laval.
The term "microbe" is first used.
1879 Flemming discovers chromatin, the rod-like structures inside the cell nucleus that later come to be called "chromosomes."
1883 The first rabies vaccine is developed.
1888 The chromosome is discovered by Waldyer.
AD: First Half of the 20th Century
1902 The term "immunology" first appears.
1906 The term "genetics" is introduced.
1907 The first in vivo culture of animal cells is reported.
1909 Genes are linked with hereditary disorders.
1911 The first cancer-causing virus is discovered by Rous.
1914 Bacteria are used to treat sewage for the first time in Manchester, England.
1915 Phages, or bacterial viruses, are discovered.
1919 The word "biotechnology" is first used by a Hungarian agricultural engineer.
1920 The human growth hormone is discovered by Evans and Long.
1927 Muller discovers that X-rays cause mutation.
1928 Fleming discovers penicillin, the first antibiotic.
1938 The term "molecular biology" is coined.
1941 The term "genetic engineering" is first used by a Danish microbiologist.
1942 The electron microscope is used to identify and characterize a bacteriophage- a virus that infects bacteria.
1943 Avery demonstrates that DNA is the "transforming factor" and is the material of genes.
1944 DNA is shown to be the material substance of the gene.
1949 Pauling shows that sickle cell anemia is a "molecular disease" resulting from a mutation.
1950 to 1960
1951 McClintock discovers transposable elements, or "jumping genes," in corn.
1953 Watson and Crick reveal the three-dimensional structure of DNA.
1954 Cell-culturing techniques are developed.
1955 An enzyme involved in the synthesis of a nucleic acid is isolated for the first time.
1956 The fermentation process is perfected in Japan.
Kornberg discovers the enzyme DNA polymerase I, leading to an understanding of how DNA is replicated.
1957 Sickle cell anemia is shown to occur due to a change of a single amino acid.
1960 Exploiting base pairing, hybrid DNA-RNA molecules are created.
Messenger RNA is discovered.
1961 The genetic code is understood for the first time.
1964 The existence of reverse transcriptase (RT) is predicted.
1967 The first automatic protein sequencer is perfected.
1969 An enzyme is synthesized in vitro for the first time.
1970s
1970 Specific restriction nucleases are identified, opening the way for gene cloning.
RT is discovered independently in murine and avian retroviruses.
1971 RT is shown to have ribonuclease H (Rnase H) activity.
1972 The DNA composition of humans is discovered to be 99% similar to that of chimpanzees and gorillas.
Purified RT is first used to synthesize cDNA from purified mRNA in vitro.
1973 Cohen and Boyer perform the first successful recombinant DNA experiment, using bacterial genes.
1974 The National Institute of Health forms a Recombinant DNA Advisory Committee to oversee recombinant genetic research.
1975 Colony hybridization and Southern blotting are developed for detecting specific DNA sequences.
The first monoclonal antibodies are produced.
1976 The tools of recombinant DNA are first applied to a human inherited disorder.
Molecular hybridization is used for the prenatal diagnosis of alpha thalassemia.
Yeast genes are expressed in E. coli bacteria.
1977 Genetically engineered bacteria are used to synthesize human growth protein.
1978 North Carolina scientists Hutchinson and Edgell show it is possible to introduce specific mutations at specific sites in a DNA molecule.
1979 The first monoclonal antibodies are produced.
1980s
1980 The U.S. Supreme Court, in the landmark case Diamond v. Chakrabarty, approves the principle of patenting genetically engineered life forms.
The U.S. patent for gene cloning is awarded to Cohen and Boyer.
1981 The North Carolina Biotechnology Center is created by the state's General Assembly as the nation's first state-sponsored initiative to develop biotechnology. Thirty-five other states follow with biotechnology centers of various kinds.
The first gene-synthesizing machines are developed.
The first genetically engineered plant is reported.
Mice are successfully cloned.
1982 Humulin, Genentech's human insulin drug produced by genetically engineered bacteria for the treatment of diabetes, is the first biotech drug to be approved by the Food and Drug Administration.
1983 The Polymerase Chain Reaction (PCR) technique is conceived. PCR, which uses heat and enzymes to make unlimited copies of genes and gene fragments, later becomes a major tool in biotech research and product development worldwide.
The first genetic transformation of plant cells by TI plasmids is performed.
The first artificial chromosome is synthesized.
The first genetic markers for specific inherited diseases are found.
Efficient methods are developed to synthesize double-stranded DNA from first-strand cDNA involving minimal loss of sequence information.
1984 The DNA fingerprinting technique is developed.
The first genetically engineered vaccine is developed.
Chiron clones and sequences the entire genome of the HIV virus.
1985 Fully active murine RT is cloned and overexpressed in E. coli.
1986 The first field tests of genetically engineered plants (tobacco) are conducted.
Ortho Biotech's Orthoclone OKT3, used to fight kidney transplant rejection, is approved as the first monoclonal antibody treatment.
The first biotech-derived interferon drugs for the treatment of cancer, Biogen's Intron A and Genentech's Roferon A, are approved by the FDA. In 1988, the drugs are used to treat Kaposi's sarcoma, a complication of AIDS.
The first genetically engineered human vaccine, Chiron's Recombivax HB, is approved for the prevention of hepatitis B.
1987 Humatrope is developed for treating human growth hormone deficiency.
Advanced Genetic Sciences' Frostban, a genetically altered bacterium that inhibits frost formation on crop plants, is field tested on strawberry and potato plants in California, the first authorized outdoor tests of an engineered bacterium.
Genentech's tissue plasminogen activator (tPA), sold as Activase, is approved as a treatment for heart attacks.
Reverse transcription and PCR are combined to amplify mRNA sequences.
Cloned murine RT is engineered to maintain polymerase and eliminate Rnase H activity.
1988 Congress funds the Human Genome Project, a massive effort to map and sequence the human genetic code as well as the genomes of other species.
1989 Amgen's Epogen is approved for the treatment of renal disease anemia.
Microorganisms are used to clean up the Exxon Valdez oil spill.
The gene responsible for cystic fibrosis is discovered.
1990s
1990 The first federally approved gene therapy treatment is performed successfully on a 4-yearold girl suffering from an immune disorder.
1991 Amgen develops Neupogen, the first of a new class of drugs called colony stimulating factors, for the treatment of low white blood cells in chemotherapy patients.
Immunex's Leukine, used to replenish white blood counts after bone marrow transplants, is approved.
Genzyme's Ceredase is approved for the treatment of Gaucher's disease.
1992 The three-dimensional structure of HIV RT is elucidated.
Recombinate, developed by Genetics Institute and used in the treatment of hemophilia A, becomes the first genetically engineered blood clotting factor approved in the U.S. Chiron's Proleukin is approved for the treatment of renal cell cancer.
1993 Chiron's Betaseron is approved as the first treatment for multiple sclerosis in 20 years.
The FDA declares that genetically engineered foods are "not inherently dangerous" and do not require special regulation.
The Biotechnology Industry Organization (BIO) is created by merging two smaller trade associations.
1994 Genentech's Nutropin is approved for the treatment of growth hormone deficiency.
The first breast cancer gene is discovered.
Calgene's Flavr Savr tomato, engineered to resist rotting, is approved for sale.
1995 The first baboon-to-human bone marrow transplant is performed on an AIDS patient.
The first full gene sequence of a living organism other than a virus is completed for the bacterium Hemophilus influenzae.
The three-dimensional structure of a catalytically active fragment of murine RT is elucidated.
1996 Biogen's Avonex is approved for the treatment of multiple sclerosis. The company builds a $50 million plant in Research Triangle Park, N.C., to manufacture the recombinant interferon drug.
Scottish scientists clone identical lambs from early embryonic sheep.
1997 Scottish scientists report cloning a sheep, using DNA from adult sheep cells.
A group of Oregon researchers claims to have cloned two Rhesus monkeys.
A new DNA technique combines PCR, DNA chips, and a computer program, providing a new tool in the search for disease-causing genes.
1998 University of Hawaii scientists clone three generations of mice from nuclei of adult ovarian cumulus cells.
Human skin is produced in vitro.
Embryonic stem cells are used to regenerate tissue and create disorders mimicking diseases.
The first complete animal genome for the elegans worm is sequenced.
A rough draft of the human genome map is produced, showing the locations of more than 30,000 genes.
Cloned vain RT with fully active polymerase and minimized Rnase H activity is engineered.
The Biotechnology Institute is founded by BIO as an independent national, 501(c)(3) education organization with an independent Board of Trustees.
1999 The complete genetic code of the human chromosome is first deciphered.
The rising tide of public opinion in Europe brings biotech food into the spotlight.
2000 and Beyond
2000 A rough draft of the human genome is completed by Celera Genomics and the Human Genome Project.
Pigs are the next animal cloned by researchers, hopefully to help produce organs for human transplant.
"Golden Rice," modified to make vitamin A, promises to help third-world countries alleviate blindness.
The 2.18 million base pairs of the commonest cause of bacterial meningitis, Neisseria meningitidis, are identified.
2001 The sequence of the human genome is published in Science and Nature, making it possible for researchers all over the world to begin developing treatments.
2002 Scientists complete the draft sequence of the most important pathogen of rice, a fungus that destroys enough rice to feed 60 million people annually. By combining an understanding of the genomes of the fungus and rice, scientists will elucidate the molecular basis of the interactions between the plant and pathogen.
2003 Dolly, the cloned sheep that made headlines in 1997, is euthanized after developing progressive lung disease. Dolly was the first successful clone of a mammal.
BC
1750 The Sumerians brew beer.
500 The Chinese use moldy soybean curds as an antibiotic to treat boils.
250 The Greeks practice crop rotation to maximize soil fertility.
100 Powdered chrysanthemum is used in China as an insecticide.
AD: Before the 20th Century
1590 The microscope is invented by Janssen.
1663 Cells are first described by Hooke.
1675 Leeuwenhoek discovers protozoa and bacteria.
1797 Jenner inoculates a child with a viral vaccine to protect him from smallpox.
1802 The word "biology" first appears.
1824 Dutrochet discovers that tissue is composed of living cells.
1830 Proteins are discovered.
1833 The cell nucleus is discovered.
The first enzymes are isolated.
1855 The Escherichia coli bacterium is discovered. It later becomes a major research, development, and production tool for biotechnology.
Pasteur begins working with yeast, eventually proving they are living organisms.
1863 Mendel, in his study of peas, discovers that traits were transmitted from parents to progeny by discrete, independent units, later called genes. His observations lay the groundwork for the field of genetics.
1869 Miescher discovers DNA in the sperm of trout.
1877 A technique for staining and identifying bacteria is developed by Koch.
1878 The first centrifuge is developed by Laval.
The term "microbe" is first used.
1879 Flemming discovers chromatin, the rod-like structures inside the cell nucleus that later come to be called "chromosomes."
1883 The first rabies vaccine is developed.
1888 The chromosome is discovered by Waldyer.
AD: First Half of the 20th Century
1902 The term "immunology" first appears.
1906 The term "genetics" is introduced.
1907 The first in vivo culture of animal cells is reported.
1909 Genes are linked with hereditary disorders.
1911 The first cancer-causing virus is discovered by Rous.
1914 Bacteria are used to treat sewage for the first time in Manchester, England.
1915 Phages, or bacterial viruses, are discovered.
1919 The word "biotechnology" is first used by a Hungarian agricultural engineer.
1920 The human growth hormone is discovered by Evans and Long.
1927 Muller discovers that X-rays cause mutation.
1928 Fleming discovers penicillin, the first antibiotic.
1938 The term "molecular biology" is coined.
1941 The term "genetic engineering" is first used by a Danish microbiologist.
1942 The electron microscope is used to identify and characterize a bacteriophage- a virus that infects bacteria.
1943 Avery demonstrates that DNA is the "transforming factor" and is the material of genes.
1944 DNA is shown to be the material substance of the gene.
1949 Pauling shows that sickle cell anemia is a "molecular disease" resulting from a mutation.
1950 to 1960
1951 McClintock discovers transposable elements, or "jumping genes," in corn.
1953 Watson and Crick reveal the three-dimensional structure of DNA.
1954 Cell-culturing techniques are developed.
1955 An enzyme involved in the synthesis of a nucleic acid is isolated for the first time.
1956 The fermentation process is perfected in Japan.
Kornberg discovers the enzyme DNA polymerase I, leading to an understanding of how DNA is replicated.
1957 Sickle cell anemia is shown to occur due to a change of a single amino acid.
1960 Exploiting base pairing, hybrid DNA-RNA molecules are created.
Messenger RNA is discovered.
1961 The genetic code is understood for the first time.
1964 The existence of reverse transcriptase (RT) is predicted.
1967 The first automatic protein sequencer is perfected.
1969 An enzyme is synthesized in vitro for the first time.
1970s
1970 Specific restriction nucleases are identified, opening the way for gene cloning.
RT is discovered independently in murine and avian retroviruses.
1971 RT is shown to have ribonuclease H (Rnase H) activity.
1972 The DNA composition of humans is discovered to be 99% similar to that of chimpanzees and gorillas.
Purified RT is first used to synthesize cDNA from purified mRNA in vitro.
1973 Cohen and Boyer perform the first successful recombinant DNA experiment, using bacterial genes.
1974 The National Institute of Health forms a Recombinant DNA Advisory Committee to oversee recombinant genetic research.
1975 Colony hybridization and Southern blotting are developed for detecting specific DNA sequences.
The first monoclonal antibodies are produced.
1976 The tools of recombinant DNA are first applied to a human inherited disorder.
Molecular hybridization is used for the prenatal diagnosis of alpha thalassemia.
Yeast genes are expressed in E. coli bacteria.
1977 Genetically engineered bacteria are used to synthesize human growth protein.
1978 North Carolina scientists Hutchinson and Edgell show it is possible to introduce specific mutations at specific sites in a DNA molecule.
1979 The first monoclonal antibodies are produced.
1980s
1980 The U.S. Supreme Court, in the landmark case Diamond v. Chakrabarty, approves the principle of patenting genetically engineered life forms.
The U.S. patent for gene cloning is awarded to Cohen and Boyer.
1981 The North Carolina Biotechnology Center is created by the state's General Assembly as the nation's first state-sponsored initiative to develop biotechnology. Thirty-five other states follow with biotechnology centers of various kinds.
The first gene-synthesizing machines are developed.
The first genetically engineered plant is reported.
Mice are successfully cloned.
1982 Humulin, Genentech's human insulin drug produced by genetically engineered bacteria for the treatment of diabetes, is the first biotech drug to be approved by the Food and Drug Administration.
1983 The Polymerase Chain Reaction (PCR) technique is conceived. PCR, which uses heat and enzymes to make unlimited copies of genes and gene fragments, later becomes a major tool in biotech research and product development worldwide.
The first genetic transformation of plant cells by TI plasmids is performed.
The first artificial chromosome is synthesized.
The first genetic markers for specific inherited diseases are found.
Efficient methods are developed to synthesize double-stranded DNA from first-strand cDNA involving minimal loss of sequence information.
1984 The DNA fingerprinting technique is developed.
The first genetically engineered vaccine is developed.
Chiron clones and sequences the entire genome of the HIV virus.
1985 Fully active murine RT is cloned and overexpressed in E. coli.
1986 The first field tests of genetically engineered plants (tobacco) are conducted.
Ortho Biotech's Orthoclone OKT3, used to fight kidney transplant rejection, is approved as the first monoclonal antibody treatment.
The first biotech-derived interferon drugs for the treatment of cancer, Biogen's Intron A and Genentech's Roferon A, are approved by the FDA. In 1988, the drugs are used to treat Kaposi's sarcoma, a complication of AIDS.
The first genetically engineered human vaccine, Chiron's Recombivax HB, is approved for the prevention of hepatitis B.
1987 Humatrope is developed for treating human growth hormone deficiency.
Advanced Genetic Sciences' Frostban, a genetically altered bacterium that inhibits frost formation on crop plants, is field tested on strawberry and potato plants in California, the first authorized outdoor tests of an engineered bacterium.
Genentech's tissue plasminogen activator (tPA), sold as Activase, is approved as a treatment for heart attacks.
Reverse transcription and PCR are combined to amplify mRNA sequences.
Cloned murine RT is engineered to maintain polymerase and eliminate Rnase H activity.
1988 Congress funds the Human Genome Project, a massive effort to map and sequence the human genetic code as well as the genomes of other species.
1989 Amgen's Epogen is approved for the treatment of renal disease anemia.
Microorganisms are used to clean up the Exxon Valdez oil spill.
The gene responsible for cystic fibrosis is discovered.
1990s
1990 The first federally approved gene therapy treatment is performed successfully on a 4-yearold girl suffering from an immune disorder.
1991 Amgen develops Neupogen, the first of a new class of drugs called colony stimulating factors, for the treatment of low white blood cells in chemotherapy patients.
Immunex's Leukine, used to replenish white blood counts after bone marrow transplants, is approved.
Genzyme's Ceredase is approved for the treatment of Gaucher's disease.
1992 The three-dimensional structure of HIV RT is elucidated.
Recombinate, developed by Genetics Institute and used in the treatment of hemophilia A, becomes the first genetically engineered blood clotting factor approved in the U.S. Chiron's Proleukin is approved for the treatment of renal cell cancer.
1993 Chiron's Betaseron is approved as the first treatment for multiple sclerosis in 20 years.
The FDA declares that genetically engineered foods are "not inherently dangerous" and do not require special regulation.
The Biotechnology Industry Organization (BIO) is created by merging two smaller trade associations.
1994 Genentech's Nutropin is approved for the treatment of growth hormone deficiency.
The first breast cancer gene is discovered.
Calgene's Flavr Savr tomato, engineered to resist rotting, is approved for sale.
1995 The first baboon-to-human bone marrow transplant is performed on an AIDS patient.
The first full gene sequence of a living organism other than a virus is completed for the bacterium Hemophilus influenzae.
The three-dimensional structure of a catalytically active fragment of murine RT is elucidated.
1996 Biogen's Avonex is approved for the treatment of multiple sclerosis. The company builds a $50 million plant in Research Triangle Park, N.C., to manufacture the recombinant interferon drug.
Scottish scientists clone identical lambs from early embryonic sheep.
1997 Scottish scientists report cloning a sheep, using DNA from adult sheep cells.
A group of Oregon researchers claims to have cloned two Rhesus monkeys.
A new DNA technique combines PCR, DNA chips, and a computer program, providing a new tool in the search for disease-causing genes.
1998 University of Hawaii scientists clone three generations of mice from nuclei of adult ovarian cumulus cells.
Human skin is produced in vitro.
Embryonic stem cells are used to regenerate tissue and create disorders mimicking diseases.
The first complete animal genome for the elegans worm is sequenced.
A rough draft of the human genome map is produced, showing the locations of more than 30,000 genes.
Cloned vain RT with fully active polymerase and minimized Rnase H activity is engineered.
The Biotechnology Institute is founded by BIO as an independent national, 501(c)(3) education organization with an independent Board of Trustees.
1999 The complete genetic code of the human chromosome is first deciphered.
The rising tide of public opinion in Europe brings biotech food into the spotlight.
2000 and Beyond
2000 A rough draft of the human genome is completed by Celera Genomics and the Human Genome Project.
Pigs are the next animal cloned by researchers, hopefully to help produce organs for human transplant.
"Golden Rice," modified to make vitamin A, promises to help third-world countries alleviate blindness.
The 2.18 million base pairs of the commonest cause of bacterial meningitis, Neisseria meningitidis, are identified.
2001 The sequence of the human genome is published in Science and Nature, making it possible for researchers all over the world to begin developing treatments.
2002 Scientists complete the draft sequence of the most important pathogen of rice, a fungus that destroys enough rice to feed 60 million people annually. By combining an understanding of the genomes of the fungus and rice, scientists will elucidate the molecular basis of the interactions between the plant and pathogen.
2003 Dolly, the cloned sheep that made headlines in 1997, is euthanized after developing progressive lung disease. Dolly was the first successful clone of a mammal.
What is Biotechnology?
What is Biotechnology?
The definition of biotechnology varies, but a simple definition is the use of organisms by man. One example of biotechnology is cloning. We have been cloning plants for centuries. Each time a leaf is excised from a violet plant and placed in soil to grow a new plant, cloning has occurred. Today, we are not only doing the physical manipulation at the visual level but also on the molecular level. In modern or molecular biotechnology, we physically select the desired characteristic at the molecular level and add it to the organism's genetic makeup.
Biotechnology is the science for this century. With its advances, we are on the first part of a great journey. Humans have expanded their understanding of the biosphere by journeying into space and exploring the depths of the ocean. We have not only been able to look at the surrounding universe and the depths below with the advancement of tools and techniques, but we also have been able to live there. The advancement tools and techniques is now allowing us to look at the universe of atoms. Biotechnology is utilizing the sciences of biology, chemistry, physics, engineering, computers, and information technology to develop tools and products that hold great promise and concern. Humans have always been "manipulating" organisms to their advantage, but now we are able to manipulate life and materials at the atomic level through nanotechnology.
The two schools of thought about what biotechnology is can elicit much debate. Both use organisms to help man. Whereas modern biotechnology manipulates the genes of organisms and inserts them into other organisms to acquire the desired trait, traditional biotechnology uses the processes of organisms, such as fermentation.
Adapted with permission from Shoestring Biotechnology: Budget-Oriented High Quality Biotechnology Laboratories for Two-Year and High School co-published by the National Association of Biology Teachers and the Biotechnology Institute.
The definition of biotechnology varies, but a simple definition is the use of organisms by man. One example of biotechnology is cloning. We have been cloning plants for centuries. Each time a leaf is excised from a violet plant and placed in soil to grow a new plant, cloning has occurred. Today, we are not only doing the physical manipulation at the visual level but also on the molecular level. In modern or molecular biotechnology, we physically select the desired characteristic at the molecular level and add it to the organism's genetic makeup.
Biotechnology is the science for this century. With its advances, we are on the first part of a great journey. Humans have expanded their understanding of the biosphere by journeying into space and exploring the depths of the ocean. We have not only been able to look at the surrounding universe and the depths below with the advancement of tools and techniques, but we also have been able to live there. The advancement tools and techniques is now allowing us to look at the universe of atoms. Biotechnology is utilizing the sciences of biology, chemistry, physics, engineering, computers, and information technology to develop tools and products that hold great promise and concern. Humans have always been "manipulating" organisms to their advantage, but now we are able to manipulate life and materials at the atomic level through nanotechnology.
The two schools of thought about what biotechnology is can elicit much debate. Both use organisms to help man. Whereas modern biotechnology manipulates the genes of organisms and inserts them into other organisms to acquire the desired trait, traditional biotechnology uses the processes of organisms, such as fermentation.
Adapted with permission from Shoestring Biotechnology: Budget-Oriented High Quality Biotechnology Laboratories for Two-Year and High School co-published by the National Association of Biology Teachers and the Biotechnology Institute.
Subscribe to:
Comments (Atom)